
IRB Information for Investigators
Information about writing and submitting a research application to the IRB, as well as information about post-approval monitoring of IRB-approved research.
Preparing Your Application
What are the obligations of Principal Investigators (PIs) who want to conduct research at Carilion Clinic?
The guidance linked below outlines the responsibilities for PIs who are interested in conducing Human Subjects Research that is overseen by Carilion Clinic's local IRB.
What should be included in a complete research protocol?
Carilion IRB does not require a separate protocol document in addition to the IRB application. The IRB application includes all protocol details. The link below provides guidance on what investigators should prepare to develop for a complete research protocol.
What does the IRB need to know about my plan to recruit and enroll participants?
The link below offers guidance for the recruitment and enrollment of potential human subjects into research activities and for the use of protected health information (PHI) as it relates to research.
Regarding PHI, does the Health Insurance Portability and Accountability Act (HIPAA) have anything to do with research?
HIPAA has requirements in addition to the ethical and regulatory protections for human subjects research. The link below provides guidance to anyone who conducts research, assists in the performance of research or otherwise uses or discloses PHI in connection with research activities at Carilion Clinic.
What is a data safety monitoring plan and when do I need to convene a Data Safety Monitoring Board?
Data safety monitoring is necessary for all greater-than-minimal-risk studies. Some research studies will require the involvement of a Data Safety Monitoring Board as a part of the data safety monitoring plan. The guidance below outlines considerations for designing a data safety monitoring plan and defines the composition, structure and responsibility of a Data Safety Monitoring Board.
Data Safety Monitoring Plan and Data Safety Monitoring Board
What do I need to include in an IRB application to develop a research repository or database?
Receiving or accessing identifiable private information or identifiable specimens for research purposes constitutes human subjects research. If you are planning to collect and disburse specimens/data at Carilion Clinic for research or operate a repository/database using specimens, then this activity is subject to oversight by the Carilion Clinic IRB. The link below provides guidance for the establishment of a repository or database of specimens or data for research and the disbursement of the stored data/specimens to researchers.
Research Involving the Collection of Specimens or Data for a Repository or Database
I'm submitting a grant application and they want the study to have IRB review prior to submission. What do I do?
Grant sponsors may differ in when they expect grants to be reviewed and approved by an IRB. The process for submitting a grant application involves preparing the necessary documentation and following institutional procedures for IRB review and approval. Contact the HRPO staff at irb@carilionclinic.org for questions and to schedule a consultation regarding the IRB review process.
Do the National Institutes of Health (NIH) requirements for data management and sharing impact the development of my research protocol?
The NIH issued the Data Management and Sharing (DMS) policy, effective January 25, 2023, to promote the sharing of scientific data. The DMS policy applies to all research funded or conducted in whole or in part by the NIH that results in the generation of scientific data.
This includes research funded or conducted by extramural grants, contracts, intramural research projects or other funding agreements, regardless of NIH funding level or funding mechanism. The DMS policy does not apply to research and other activities that do not generate scientific data, including training, infrastructure development and non-research activities.
Data Management and Sharing Policy
Data Management and Sharing Sample Template
What do I need to know about IRB approval for international research?
The link below outlines the criteria used by the Carilion Clinic IRB in the review of international research.
PRIS3M Submission System
Required Education for Investigators
All individuals engaged in human subjects research at the Carilion Clinic must take the Carilion-specific CITI Training human participant protections training before their research can be approved by the IRB.
Each CITI curriculum consists of a series of required modules and each module has a short quiz. You must pass each quiz with a minimum score of 80%. A running tally is compiled in the grade book. If you want to improve a score, you may repeat any quiz in which you did not score 100% correct by selecting the View This Module Again tab.
Investigator and Key Study Personnel Guidance
Biomedical Researchers
This is required for all Investigators (i.e., Primary Investigators and co-investigators) who are responsible for the project as a whole and all project staff who have direct contact with human participants (e.g., for subject recruitment and data collection) or who have access to information that links participants’ names with their data. This is an entry-level course suitable for investigators and staff conducting biomedical research with human subjects.
A Basic course must always be taken before a Refresher course can be accepted. If you do not have a record of previous course completion or HRPO staff cannot find records via CITI, you will not receive previous credit.
Three years after the completion of the Basic course, investigators and staff must take the Refresher 1 module in order to participate in new studies. After six years, investigators and staff must take the Refresher 2 module in order to participate in new studies.
Good Clinical Practice (GCP)
This course, in addition to the Human Subject Research Biomedical Investigators course, is required for all research team members who are responsible for the conduct, management and oversight of NIH-funded applicable clinical trials or FDA-regulated research (research involving drugs, devices or biologics, whether FDA approved or experimental).
GCP training must be refreshed at least every three years to remain current with regulations, standards and guidelines.
Conflict of Interest Mini-Course
This must be completed by researchers and staff who conduct research with external funding or support.
The mini-course must be taken once every four years.
Note: The Social Behavioral Human Subjects Course has been removed and is no longer an option. You may add supplemental courses that are applicable to your research to the Biomedical Researchers course.
All required modules must be completed in order to receive credit for the training course. Upon completion of a required module, it is your responsibility to print or download a Course Completion Report as evidence that you have met the IRB training and education requirement. The IRB may request a copy of your completion report with your IRB submission. Email irb@carilionclinic.org if you have any questions.
Categories of Review
How is research defined by the federal regulations?
The Office of Human Research Protections (OHRP) defines research as a systematic investigation, including research development, testing and evaluation, that leads to generalizable knowledge.
A human subject is a living individual from whom an investigator obtains information or biospecimens through intervention or interaction with the individual and uses, studies or analyzes the information or biospecimens; or obtains, uses, studies, analyzes or generates identifiable private information or identifiable biospecimens.
Determining level of review
The IRB assigns each application to an appropriate level of review:
- Not Human Subjects Research
- Quality Improvement/Quality Assurance
- Exempt
- Expedited
- Full Board
How does the IRB determine the level of review for my application?
The guidance linked below describes how the Carilion Clinic IRB differentiates human subjects research from non-human subjects research, and the level of review needed for each proposal.
How do I know if my proposal is human subjects research or not human subjects research?
The guidance linked below will help investigators determine whether a project is human subjects research, not human subjects research or a quality assurance/quality improvement project. The guidance also includes considerations for case reports.
Not Human Subjects Research, QA/QI Submissions, Case Reports
What type of research is considered exempt?
Research that may qualify under this category generally involves education, behavioral or social science studies that present little or no risk to the study subjects.
Click/tap the link below for more guidance and additional types of research that may qualify for exempt review.
What type of research may qualify for expedited review?
Research activities that present no more than minimal risk to human subjects and involve only procedures that are outlined in the link below. Please note that exempt categories do not permit FDA-regulated studies.
When will my proposal need to be reviewed by the full board?
Full board review of research is conducted when the research does not meet the criteria for exemption of review or expedited review or otherwise determined to necessitate review at a convened meeting of an IRB committee. Click/tap below for guidance on full board review.
IRB Application Templates
PRIS3M Application Templates
IRB Application templates with branching options are provided as examples only. Submissions to the Carilion Clinic IRB are required to be submitted in PRIS3M submission system. Please contact our office at irb@carilionclinic.org with any questions.
New Submissions
How To Submit a Study to the IRB
All new submissions must come through the PRIS3M online submission system. To access the PRIS3M system for the first time, sign in with your Carilion Clinic Active Directory user name and password and an account will be created for you. If you do not have an active directory user name and password, please contact the Primary Investigator for the study and/or the Department Chair.
Before getting started, view the video Signing Into the PRIS3M System linked below.
Click the PRIS3M Submission System button below for access to the online submission system.
What types of applications are supported in the PRIS3M system?
The PRIS3M application supports multiple types of research applications, including:
- Human Subjects Research Study
- Determination of Human Subjects Research
- Quality Assurance/Quality Improvement
- Case Reports
- Use of De-Identified Data
- Establishing a Prospective Data or Specimens Research Repository
- Humanitarian Use Device (non-research use)
- Expanded Access or Compassionate Use
- Single Patient Emergency Use
- Preparatory to Research Application
- IRB Grant Review (only for preliminary approval if required by funder)
- Requesting Carilion Clinic Rely on Another IRB of Record (WCG IRB, NCI IRB, VT, UVA, Advarra, etc.)
How do I know where my study is in the PRIS3M system?
Study Status in PRIS3M outlines the status of studies in the PRIS3M system to assist investigators in understanding the way that studies progress through the system.
User Guides
Find PRIS3M user guides, training materials, and step-by-step instructions on Inside Carilion. Resources cover common workflows for investigators, study teams, department leaders, and IRB members.
Note: Inside Carilion is available to Carilion employees and affiliated users and requires login.
Training Video
This training video shows you how to navigate a stipulation request to a change/update form.
Changes and Updates
We need to make some changes to our approved study. What do we need to know?
Changes that need to take place in a research study must be approved by the IRB before those changes may be implemented. The link below outlines guidance on the process for notifying the IRB about any changes or updates to approved research projects.
Examples of changes that need to be submitted to the IRB before implementation include:
- Study procedures
- Consent document/process
- Study documents
- Changes in personnel
- Inclusion of additional participants
- Increase in length of study
- Identification of new risks
- Closure to accrual of new subjects (analysis of identifiable data ongoing)
Additional examples and guidance can be found through the link below
How to Notify IRB of Changes/Updates to Research Projects
User Guide: How to Submit a Post-Approval Change/Update Submission in PRIS3M
Continuing Review/Annual Check In
Continuing Review
Federal regulations mandate that a research protocol must be reviewed by the IRB at intervals appropriate to the degree of risk, but not less than at least once every 12 months for the duration of the study. Continuing review must be substantive and meaningful, and it must address any new information or changes that relate to risk/discomfort, benefits, safeguards for participants, and informed consent to assure that all criteria for approval specified under 45 CFR 46.111 and/or 21 CFR 56.111 are satisfied.
Expedited studies that are FDA regulated require a continuing review as well as studies that were approved before the 2018 Common Rule Change. The IRB may require a continuing review for any expedited study at its discretion.
Annual Check-In
The IRB may determine that an Annual Check-in review process is appropriate for studies that are approved according to expedited criteria and that do not require a full continuing review. These studies are minimal risk and not FDA regulated.
The link below provides guidance for the continuing review and study check-in process.
Continuing Review and Check In
User Guide: Submitting a Continuing Review, Annual Check-in, PRI or a Conclusion Form in PRIS3M
Promptly Reportable Information
What is an adverse event and when do I need to report it to the IRB?
It is important to be aware that certain events must be reported to the IRB within a specific time frame. The link below clarifies when unanticipated problems, including adverse events, must be reported to the Carilion Institutional Review Board (IRB), and outlines the process for reporting them.
What constitutes non-compliance?
Below you will find guidance outlining procedures for handling non-compliance with regard to research activities.
Conduct of Research: Non-Compliance, Suspension, Termination, Unanticipated Problem
What should occur when a complaint is received?
Below you will find guidance outlining procedures for handling complaints from a participant or any other source.
Procedure for Complaints regarding Human Subjects Research
What is research misconduct and how is it addressed by the IRB?
Research misconduct is defined as the fabrication, falsification or plagiarism in proposing, performing or even reviewing research, or in reporting research results. The link below further delimits what constitutes research misconduct.
User Guide: Submitting a Continuing Review, Annual Check-in, PRI or a Conclusion Form in PRIS3M
Study Completion: Ending IRB Oversight
What do I need to do when I want to close a study?
IRB oversight must continue through the data analysis phase if data analysis includes identifiable data. When all study conduct is concluded, including the review of identifiable data, the investigator must submit a conclusion form through the PRIS3M system. The link below offers additional guidance for study conclusion.
Study Conclusion and the End of IRB Oversight
I concluded a study, but now I need to re-open it. What should I do?
Once a study has been concluded, no further activity may take place. There are occasions when a study team will request to re-open a study. The linked guidance provides information for the process of re-opening a closed study.
Re-Opening a Study After Conclusion
User Guide: Submitting a Continuing Review, Annual Check-in, PRI or a Conclusion Form in PRIS3M
Relying on External IRBs and Collaborations
About Carilion Clinic IRB Reliance Agreements
An IRB reliance agreement is a formalized agreement that allows an institution engaged in non-exempt human research to entrust IRB review and approval to an external IRB. Detailed information about this process can be found at the link below.
What do I need to know about IRB Reliance Agreements for multi-site research?
A reliance agreement allows one IRB (IRB of Record) to review research for multiple sites, reducing duplicate oversight. Several factors determine what IRB will serve as the IRB of Record. Click on link below for overview:
Single IRB Review Reliance Agreement Overview
The link below outlines the process to request the Carilion IRB to rely on an external IRB using an established Master Agreement.
Requesting Carilion IRB to Rely on an External IRB: Established Master Agreements Only
What do I need to do if I want to collaborate on a multi-center, industry-sponsored study using the commercial IRBs Advarra or WCG?
The link below describes the process of interaction between the Carilion Clinic IRB and the Advarra or WCG IRB.
What are Carilion Clinic's IRB review fees for industry/commercially-supported research studies when Carilion is relying on the other IRB?
All new studies with industry/commercial sponsors with contracts being negotiated on or after June 1, 2023 will include the Carilion IRB fee of $500 for a local-context administrative review.
Administrative review where Carilion is the relying IRB will include the Carilion IRB fee of $500.
What do I need to do if I want to open an adult or pediatric cooperative group oncology trial approved by the National Cancer Institute Central IRB (NCI CIRB)?
The link below offers guidance for the process of interaction between the Carilion Clinic IRB and the NCI CIRB.
I think my research study will need to be reviewed by the Carilion IRB and the IRB of another institution. What do I need to know?
In some cases research protocols may require the review of the Carilion Clinic IRB and the IRB of another institution. In all of these instances, a written memorandum of understanding that is executed between the Carilion IRB and the IRB of the outside institution will offer clarity to the IRB oversight process. Click/tap below to learn more about reciprocal IRB review and reliance agreements.
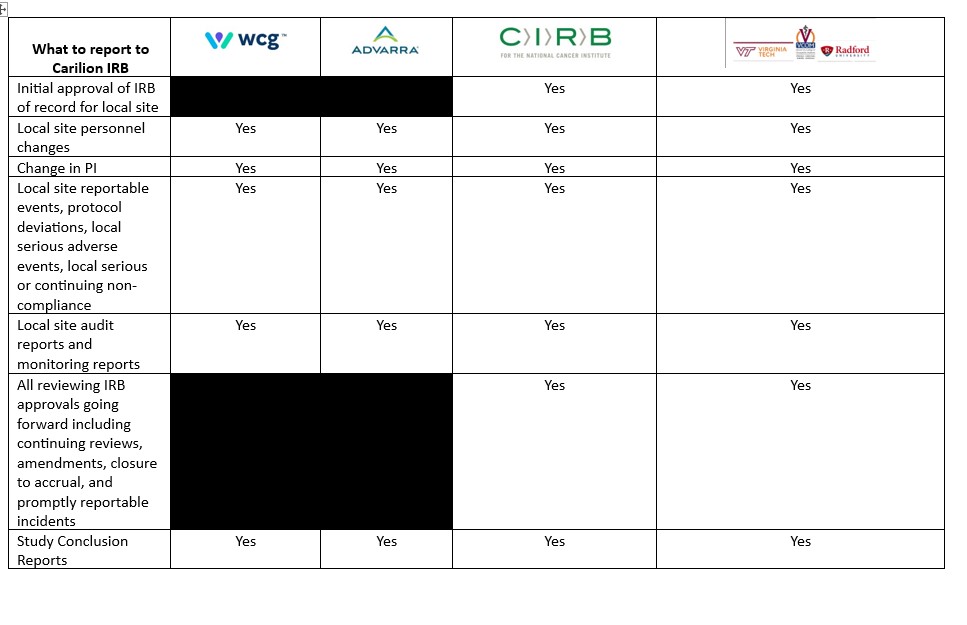
Reporting Requirements for Request to Rely Studies
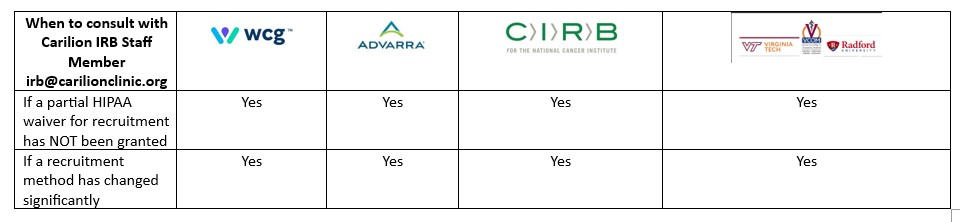
When to consult with the Carilion IRB
Vulnerable Populations
Who are considered to be especially vulnerable in a research setting, and what do I need to know to include these individuals in research?
Certain groups of human subjects are considered to be particularly vulnerable in a research setting. The guidance linked below defines vulnerable populations and notes additional approval criteria and safeguards that must be met prior to IRB approval of research involving vulnerable individuals.
Drugs, Devices and Biologics
Emergency and Treatment Use of Investigational Drug or Biologic
When can an investigational medical device be used and when should this be communicated to the IRB?
The guidance below outlines the IRB process for review and approval of investigational medical devices, including emergency and compassionate use, as well as humanitarian use devices.
Can I use an investigational drug or biologic for emergency or treatment?
The link below outlines guidance for IRB review and approval of research involving the emergency and treatment use of investigational drugs. When reviewing the guidance, make note of the required time periods for communicating with and reporting to the IRB.
Emergency and Treatment Use of an Investigational Drug
Genetic Testing Guidance
What do I need to know about collecting and analyzing human biological samples?
Research involving human biological samples requires careful consideration for DNA, RNA and protein analysis. Study submissions must detail sample collection methods, storage procedures and analytical techniques while protecting participant rights.
See detailed guidance below for specific requirements on sample handing, analysis methods and participant protections.
Post-Approval Monitoring
The HRPO offers several post approval monitoring programs.
Research Partner Documentation Assessments (RPDAs)
RPDAs are conducted for internal quality review of studies and are intended to be a collaborative review process. RPDAs allow us to address any questions from or needs of the research team. This process also provides an opportunity to identify any issues that may become challenges as the study progresses. Our goal is to work together to problem solve and develop solutions as early as possible.
Direct or For-Cause Reviews
These are conducted at the request of the IRB, the IRB chair, the HRPO director, the Institutional Official or their designee.
Voluntary Reviews
These are conducted on request of the Principal Investigator (PI) to support self-assessment and improvement efforts by the PI and/or study team.
Human Research Protections Program Quality Assurance
These reviews are conducted periodically to track and improve overall satisfaction and institutional compliance with human research protections program requirements.
Additional Information
External Resources/Links
Carilion Clinic Resources
- MyProjectPath, a virtual assistant to navigate research and quality assurance/quality improvement processes and resources at Carilion Clinic.
- Carilion Clinic Research and Development
- Carilion Clinic Privacy Policy
External Resources
- Office for Human Research Protections (OHRP), U.S. Department of Health and Human Services
- U.S. Food and Drug Administration (FDA)
- Public Responsibility in Medicine and Research (PRIM&R)